




Neuroendocrine Tumors
Latest News
Advertisement
Latest Videos



CME Content



















































Advertisement
More News
In general, NETs are considered an “immunological desert,” but a new study showed some promise for immunotherapy.

A retrospective analysis found that surgical resection and radiotherapy are linked to superior outcomes in patients with thymic neuroendocrine tumors.
Radioguided surgery using gallium 68 dota peptides offers a highly sensitive method for detecting neuroendocrine tumors.
For patients with NETs that have metastasized to the liver, high levels of serum pancreastatin are predictive of poor outcomes after treatment with TACE.
A new study suggests that the cytokine CXCL12 could be useful as a prognostic biomarker in patients with neuroendocrine tumors of the lung.

A study on pheochromocytoma and paraganglioma associated with neurofibromatosis type 1 highlights how screening for these malignancies is important.

Researchers have developed a clinical prediction model for the metastatic potential of pheochromocytoma and paraganglioma.
Intravenous Iobenguane I 131 has been approved to treat unresectable, locally advanced, or metastatic pheochromocytoma.

The US Food and Drug Administration approved lutetium Lu 177 dotatate (Lutathera) for the treatment of somatostatin receptor–positive gastroenteropancreatic neuroendocrine tumors in adults.
Patients with gastrointestinal neuroendocrine tumors with carcinoid syndrome are more than twice as likely to have certain pre-existing diagnoses compared with patients without carcinoid syndrome, according to the results of a study.
Researchers are proposing the consideration of expanding criteria for liver debulking in pancreatic neuroendocrine tumors to include a threshold of greater than 70% debulking, intermediate grade tumors, positive margins, parenchyma-sparing resections, and extrahepatic metastases.
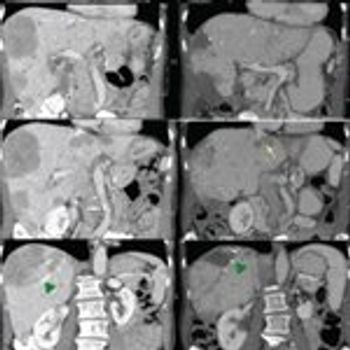
In this review, we focus on the treatment of well-differentiated early and metastatic PNETs, emphasizing current controversies, recent advances in therapy, and the multidisciplinary approach required for optimal treatment.
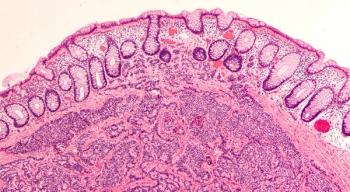
A study exploring the genetic underpinnings of the newly classified high-grade neuroendocrine carcinoma has found that it is a rare but aggressive tumor with a high frequency of BRAF mutations.
Treatment of advanced, non-functional gastrointestinal or lung neuroendocrine tumors with everolimus resulted in improved progression-free survival with no relevant differences in health-related quality of life compared with placebo, according to an analysis of the RADIANT-4 trial.
The FDA has approved telotristat ethyl (Xermelo) tablets for the treatment of patients with carcinoid syndrome diarrhea that has not responded to somatostatin analogs alone.
Everolimus improved progression-free survival by 6 to 8 months compared with placebo in patients with advanced neuroendocrine tumors of the gastrointestinal tract.
A novel drug, 177Lutetium-DOTATATE (Lutathera), significantly lowered the risk for disease progression or death among patients with previously treated, advanced midgut neuroendocrine tumors.
Small- and large-cell neuroendocrine tumors of the cervix are exceedingly rare and exceedingly aggressive.
Small intestinal “carcinoid” or well-differentiated grade 1 neuroendocrine tumors can have an insidious onset or be diagnosed serendipitously at the time of surgery, during the workup for another disorder, or during a screening test.
Nonfunctional neuroendocrine tumors of lung or gastrointestinal origin were safely and effectively treated with the mTOR inhibitor everolimus.
Patients with midgut neuroendocrine tumors had significantly delayed disease progression when treated with Lutathera compared with the current standard of care.

The US Food and Drug Administration approved lanreotide for the treatment of patients with gastroenteropancreatic neuroendocrine tumors.
In patients with advanced, unresectable NETs, there are several treatment options; which of these may be considered depends on the site of origin of the tumor.
Treatment-emergent small-cell/neuroendocrine prostate cancer is likely to become of increasing clinical relevance in the era of widespread use of potent androgen receptor–targeted therapies.
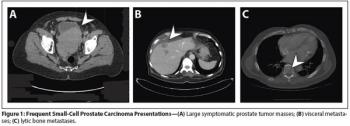
Aggressive variants of prostate cancer often take the form of neuroendocrine or small-cell carcinomas, which frequently lack androgen receptor expression and respond poorly to hormonal therapies.
Advertisement
Advertisement
Trending on CancerNetwork
1
FDA Approves RP1/Nivolumab for Progression on Anti–PD-1 Therapy in Melanoma
2
Developers Initiate Phase 2/3 Ivonescimab/EV Trial in Bladder Cancer
3
The Lowest-Ever Hazard Ratio in Multiple Myeloma Bispecific Trials?
4
Invasive Lobular Carcinoma: Diagnosis, Trials and What's Changing
5