














































It is clear that the management of adult patients with ALL is an area in which little progress has been made in the last 30 years. Given the disappointing outcomes, the field is one that lends itself to the study of the incorporation of novel agents, including monoclonal antibodies and tyrosine kinase and proteasome inhibitors, as well as to further study of allogeneic transplant.
Lymphoma
Latest News
Advertisement
Advertisement
In this review, we will discuss the management of ALL in the adult population, in the context of the recently published guidelines from the NCCN. We will focus in particular on the strides being made in salvage and targeted approaches.

A study comparing dasatinib to imatinib in patients with chronic myeloid leukemia found that the second-generation TKI dasatinib yielded better remission and molecular response rates than imatinib, but these did not result in better survival outcomes.

The FDA approved the tyrosine kinase inhibitor bosutinib (Bosulif) to treat Philadelphia chromosome–positive chronic myelogenous leukemia (CML).
A common genetic mutation in BIM appears to play a role in resistance to tyrosine kinase inhibitors in chronic myeloid leukemia (CML) and EGFR-mutated NSCLC.
The FDA has approved vincristine sulfate liposome injection (Marqibo) to treat adults with Philadelphia chromosome–negative acute lymphoblastic leukemia (ALL).
A new study finds that adding rituximab maintenance to R-CHOP induction is an effective treatment for older patients with mantle-cell lymphoma.
Ariad Pharmaceuticals has asked the FDA for an accelerated approval of the BCR-ABL inhibitor ponatinib, based on promising data from ongoing phase II trials.
Acute myeloid leukemia (AML) encompasses multiple disease entities that differ with regard to marrow morphology, cytochemistry, immunophenotype, pretreatment clinical characteristics, and treatment outcome.
Prognostic factors in acute myeloid leukemia (AML) may be subdivided into those related to patient characteristics and general health condition, and those related to characteristics of the tumor.
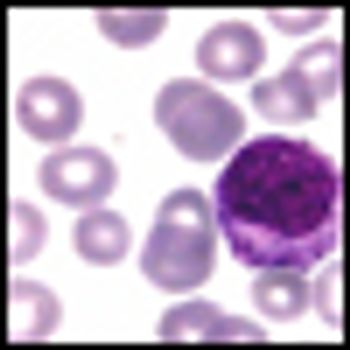
Three-year results from the ENESTnd trial show continued superiority of nilotinib over imatinib in patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) in chronic phase, according to a new paper published online ahead of print in Leukemia in June.
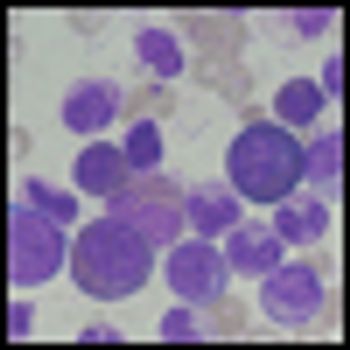
Nilotinib offers good results for patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) in chronic phase who are resistant or intolerant of imatinib, according to 48-month follow-up results of a phase II study.
Observations made in the study of the leukemias have contributed to our understanding of malignancy in general and to our ability to treat cancer patients with drugs.
The article by Drs. Stein and Tallman is an excellent summary indicating that several different approaches may lead to the cure of acute promyelocytic leukemia (APL).
The management of patients with acute promyelocytic leukemia (APL) has been transformed over the course of the last two decades following the introduction of successful molecularly targeted therapies-all-trans retinoic acid (ATRA) and arsenic trioxide (ATO)- which act in concert to induce degradation of the PMLRARα oncoprotein formed by the chromosomal translocation t(15;17)(q22;q21).

The treatment of APL in the modern era is a success of modern hematology. In this review we have attempted to plant the seeds of understanding regarding how diagnosis and treatment of APL will be pursued over the next decade.
Updated clinical data from the pivotal phase II global PACE trial of ponatinib confirm its impressive antileukemic activity in patients with chronic myeloid leukemia or Philadelphia-chromosome-positive acute lymphoblastic leukemia at all stages who are resistant or intolerant to dasatinib or nilotinib.
Bendamustine plus rituximab should be considered as the preferred first-line treatment of follicular and indolent lymphomas, and the elderly with mantle cell lymphoma.
In a phase I study the targeted drug crizotinib, a small molecule inhibitor of anaplastic lymphoma kinase and met proto oncogene, delayed or eliminated signs of tumor growth in pediatric patients with aggressive cancers.
A new study has identified independent risk factors for the development of non-Hodgkin lymphoma (NHL) including high fetal growth, older age of the mother, low birth order, and male sex. A family history of NHL in either parent or sibling was found to be the strongest risk factor.
Researchers at the University of Pennsylvania have identified a protein that could be targeted to turn off B-cell lymphomas. The protein, CD19, was found to be a major regulator of B-cell neoplastic growth driven by the MYC oncogene.
Dr. Hoppe and colleagues present a strong case supporting the use of proton therapy (PT) for the treatment of patients with Hodgkin lymphoma (HL).
Proton radiotherapy is here to stay. Despite the high initial cost, the number of proton therapy machines in the United States and elsewhere is increasing rapidly.[1] The major questions now relate to defining and optimizing their appropriate use.
If there is one disease for which patients have experienced a significant improvement in the cure rate over the past 10 years, it is diffuse large B-cell lymphoma (DLBCL).
In their article in this issue of ONCOLOGY, Nastoupil, Rose, and Flowers give a very careful assessment of the options for treating diffuse large B-cell lymphoma (DLBCL), with a focus on the importance of dose density in improving outcomes in this disease.
Advertisement
Advertisement
Trending on CancerNetwork
1
The Beginning of the “Immunotherapy Deluge” in Multiple Myeloma
2
Salt-Formulation Cabozantinib Capsules Receive FDA Tentative Approval in RCC, HCC, epNET
3
TIL Cell Therapy in HIV Positive Patient With Metastatic Melanoma: Case Report
4
Gedatolisib/Fulvestrant Earns NCCN Category 1 Listing for HR+/HER2- Breast Cancer
5